© SEQme s.r.o., 2012 - 2024. Všechna práva vyhrazena.
Právní upozornění.
Webdesign Vibes Vision
Kombinace Illumina a ONT readů – výhoda pro de novo assembly genomů
Úvodem
Ačkoli v současné době technologie Next-Generation sekvenování (NGS) generují dostatečné množství dostatečně kvalitních dat potřebných pro de novo assembly genomů organismů, u kterých zatím není známá referenční sekvence, samotný proces sestavení genomu je pro bioinformatiky stále velkou výzvou. V tomto článku se pokusíme nastínit problematiku assembly a ukázat možná řešení.
Jedna z nejrozšířenějších NGS platforem je Illumina. Její výhodou je především vysoká kvalita čtení, možnost sekvenovat v párovém nastavení, poměrně dostupná cena sekvenování a robustnost. Nevýhodou je délka čtení, která je pro tuto platformu limitující (max. 2x 300 b), a fakt, že i při vysokém pokrytí dochází k nedostatečnému nebo žádnému prosekvenování specifických oblastí genomu jako jsou GC bohaté oblasti, začátky a konce chromosomů, repetitivní oblasti atd. To má negativní vliv na proces sestavení kompletního genomu pouze na základě Illumina dat – výsledkem bývají nespojité fragmenty, tedy větší množství kratších kontigů, které není jednoduché správně složit v jeden souvislý celek. Tato „hluchá místa“ mohou být částečně překlenuta pomocí sekvenování tzv. mate-paired knihoven. Mate-paired sekvenování neodstraní mezery vzniklé během assembly, ale pomáhá uspořádat kontigy do správného pořadí.
Další možnost je doplnění assembly o dlouhé fragmenty DNA získané z jiných sekvenačních platforem. Jako jedna z vhodných technologií sekvenování dlouhých readů se jeví technologie Oxford Nanopore (ONT). Pomocí této technologie lze dle specifikací výrobce číst ready dlouhé až několik megabází, ale v běžném výstupu jde většinou o několika kilobázové ready, které pro účely assembly bohatě stačí. I když technologie Oxford Nanopore zatím zaostává za Illuminou co se kvality čtení týče, vývoj v této oblasti je celkem rychlý a neustále se zlepšuje jak po stránce technologické, tak analytické.
V principu by bylo možné pro složení genomu použít pouze tuto technologii dlouhých readů, ovšem vzhledem k chybovosti čtení by byla nutná vyšší hloubka čtení, než je tomu u sekvencí získaných technologií Illumina. Nabízí se tedy otázka, zda je možné tyto postupy spojit dohromady a získat tak co nejkvalitněji složený genom. Řešení je hned několik. Například tzv. hybridní assembly, tedy kombinace kratších readů s dobrou kvalitou čtení s dlouhými ready ideálními pro překlenutí mezer v assembly. Druhou možností je assembly založené na dlouhých readech (např. ONT) a vylepšené pomocí vysoko kvalitních krátkých readů Illumina – tzv. polishing.
V nedávné době jsme obdobnou situaci řešili – prováděli jsme de novo assembly bakteriálního genomu za využití dat jak z Illumina tak z ONT sekvenování a sestavili jsme genom vícero přístupy, abychom nalezli nejideálnější postup pro daná data.
Pro hybridní assembly byl zvolen program SPAdes Genome Assembler, což je assembler používající k assembly readů de Bruijnových grafů, a byl vyvinut pro de novo assembly malých genomů. Umožňuje zpracovávat krátké i dlouhé sekvence samostatně nebo je kombinovat mezi sebou. Druhým přístupem bylo assembly dlouhých readů za použití programu Canu, který se specializuje na assembly readů ze sekvenačních platforem PacBio a Oxford Nanopore. Canu pracuje ve třech fázích: korekce, trimming a assembly. Nekonzistentní místa assembly dlouhých readů jsou následně opravena pomocí Illumina dat v programu Pilon.
Pozn.: Všechny programy použité v tomto srovnání jsou volně dostupné, můžete je dále šířit a / nebo upravovat podle podmínek GNU General Public License verze 2.
Data
V našem případě byl složen nový bakteriální genom, u kterého byla předpokládaná velikost 3 Mb a vyšší procentuální zastoupení GC bází. K tomuto účelu byla připravena:
- Celogenomová shotgun knihovna, která byla sekvenována v nastavení 2x150 bp (Illumina),
- Genomová DNA (neštěpená) pro nanopórové sekvenování (technologie ONT). Průměrná délka readů se pohybovala kolem 10 kb.
Pro porovnání byly provedeny celkem čtyři různé postupy assembly:
- De novo assembly složené pouze z Illumina dat (SPADes)
- Hybridní de novo assembly Illumina dat v kombinaci s ONT daty (SPADes)
- De novo assembly pouze z ONT dat (Canu)
- De novo assembly z ONT dat, následný polishing Illumina daty (Canu, Pilon)
Porovnání výstupů bylo provedeno pomocí programu Quast. Statistiky byly vypočítány z kontigů >= 500bp. Sumární výstupy analýz jsou uvedeny v tabulce níže.
Výsledky
- Jako první byly hodnoceny výstupy assembly z programu SPADes při použití pouze Illumina dat. Celkově bylo složeno 50 kontigů, z toho 31 bylo kratších než 500 bp, zbylých 19 bylo zahrnuto do dalších statistik. Nejdelší složený kontig byl dlouhý 659 929 bází a celková délka assembly dosahovala 2 727 401 bází. Hodnota N50 byla 359 855 bází a L50 třem kontigům, přičemž obsah GC činil 70,63%.
- Druhý sloupec popisuje hybridní assembly, které bylo sestaveno opět programem SPADes. Celkový počet složených kontigů klesl na 12, z toho 2 kontigy byly delší než 500bp. Nejdelší kontig dosáhl délky 1 768 225 bází a celková délka assembly byla 2 735 673 bází, obsah GC 70,60%. N50 se vyšplhalo na 1 768 225 bází a L50 odpovídal jeden kontig.
- Třetí sloupec reprezentuje assembly výlučně dlouhých readů pomocí Canu assembleru. Výstupy obsahovaly jediný kontig o celkové délce 2 742 214 bází a GC složení 70,83%.
- Čtvrtý sloupec obsahuje výsledky assembly dlouhých readů získaných programem Canu a draft polishing provedeným programem Pilon. Výsledné assembly obsahuje také pouze jediný kontig, ale jeho celková délka se mírně zvýšila na 2 774 588 bází a obsah GC klesl na 70,63%.
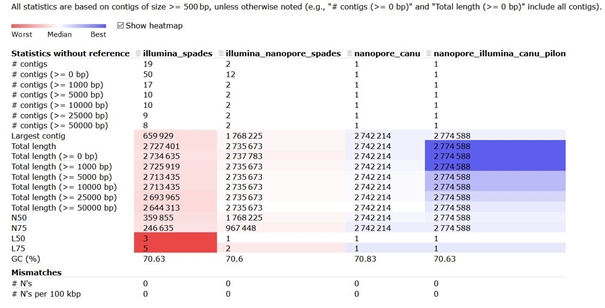
Statistické porovnání výstupů čtyř assembly - Quast.
Zhodnocení
Ve všech čtyřech případech se celková velikost assembly pohybovala okolo 2,7 Mb, což odpovídalo předpokládané velikosti zkoumaného genomu. V prvním případě (Illumina_spades) byl genom složen do několika kontigů, z čehož více než polovina byla menší než 500 bp. Nejdelší kontig odpovídal zhruba jedné čtvrtině celkové délky assembly. Pokud porovnáme výsledky s druhým výstupem (Illumina_nanopore_spades) byl vidět zajímavý rozdíl při použití dlouhých readů. V případě tohoto hybridního assembly klesl počet kontigů (přesto většina nedosahuje délky větší než 500 bp), celková délka assembly se prodloužila, ale mnohem zajímavější je délka nejdelšího kontigu, ta narostla více než 2,5x oproti assembly pouze z Illumina dat! Je tedy zřejmé, že dlouhé ready pomohly překlenout oblasti, které program SPAdes nedokázal složit pouze z krátkých sekvencí.
Naproti tomu assembly dlouhých readů složilo jeden finální kontig (nanopore_canu). Oproti předchozím výsledkům můžeme pozorovat lehce vyšší GC obsah, což může být způsobeno horší kvalitou čtení ONT readů. Poslední sloupec reprezentuje výsledky assembly dlouhých readů po následné opravě pomocí Pilonu za pomoci Illumina readů (Nanopore_illumina_canu_pilon). Tím se podařilo finální kontig ještě mírně prodloužit a upravit GC složení.
Tyto výsledky jasně naznačují, že de novo assembly vzniklé kombinací krátkých readů (Illumina dat) s dlouhými ready (ONT daty) přináší velmi přesvědčivé výsledky a pomáhá technicky zlepšit finální sestavu genomu. Ačkoli nelze z těchto výsledků se 100% jistotou říci, že byl genom složen kompletně a správně (což nelze bohužel v podstatě nikdy), přiblížili jsme se očekávaným předpokladům a běžně používané metriky v tomto případě jasně ukazují na vhodnost tohoto přístupu. Přesto nelze obecně deklarovat, že pouze metoda assembly s následným polishing je tím nejlepší přístupem. Nezbytná je následná anotace genomu a ověření výsledků in vitro. Vždy je třeba zohledňovat specifika jednotlivých druhů, celkovou velikost a komplexnost genomu. Proto doporučujeme během analýzy dat použít více postupů a kriticky zhodnotit jejich výsledky, na základě kterých by mělo dojít k rozhodnutí, který z nich je pro konkrétní data nejvhodnější.
Rozhodně je však zřejmé, že uvedená kombinace technologií ONT a Illumina, jakkoliv finančně náročnější než použití jen jedné z nich, přináší v případě de novo assembly zásadní výhodu pro jeho úspěšné sestavení. V případě, že máte v plánu obdobný projekt, doporučujeme vám, abyste včas kontaktovali naše aplikační specialisty a diskutovali s nimi jeho provedení. Shora uvedený příklad je dokladem toho, že základem úspěchu je projekt dobře naplánovat a v tom vám rádi pomůžeme!
Reference:
- Pilon - Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE. (2014)
- Canu - Koren S, Walenz BP, Berlin K, Miller JR, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Research. (2017)
- SPADes – Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology. (2012)
- Quast - Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. (2013)