Illumina technology is the so-called short read technology. It is the technology of choice if, for example, you want to analyze changes in gene expression, detect variants in (target regions of) the genome or exome, or perform shallow rather than deep screening of communities of different organisms including their functional genes. And of course in other scenarios. However, for various other applications you may rather consider long-read technology by Oxford Nanopore.
We offer and perform:
- All steps starting from project design and consulting, through DNA/RNA extraction, library preparation and sequencing using state-of-the-art instruments to standard or customized data analysis pipelines
- Solutions for large or small scale NGS projects – ShareSeq data packages
- Strict quality control on the level of the sample, the library and the data, using workflows optimized for speed and accuracy
- On-line ordering, results reporting and data delivery – all available 24/7
- Courses or workshops, also tailored to needs of your team

| You order | Sequencing by lane / flowcell | ShareSeq data package |
| What is the service suitable for? | Rather larger projects with a lower price per unit of data (eg one million reads) | Rather small to medium projects with a lower total cost |
| Available sequencing settings | Paired-end 150b, Paired-end 250b | |
| Data amount | 375 Gb and more | 1 Gb and more |
| Guarantee | The amount of data output is approximate. | If library prep is being made in our lab, we guarantee the amount of data specified for ShareSeq data packages! |
| Library preparation | We or you | |
| Library type | No limits | |
| Adapters | Compatible with standard Illumina sequencing primers. | |
| Indexes | Exclusively dual indexes. If you prepare libraries yourself and do not want / cannot use dual indexes, always contact us first. | |
| Outputs | Standard (commonly used data formats etc.). If you order a lane or flowcell, you can also get non-demultiplexed data from us (not possible with ShareSeq data packages). | |
Below is information on a few selected applications. The technological spectrum of Illumina sequencers is very extensive, in case of other applications, do not hesitate to contact us.

If you want to analyze the occurrence of microorganisms in a certain environment, metagenome sequencing is a very efficient tool of first choice. The most frequently used strategy is based on the sequencing of amplicons obtained by amplification of selected genes (long-read metagenomics) or their hypervariable regions (short-read metagenomics). Typically, these are genes for ribosomal subunits (such as 16S, 18S or ITS), but not only them. These genes or gene regions are traditionally used to identify organisms based on comparison of the obtained sequences with reference databases.
You can find the current available primer sets here. You need some other primers and cannot see them in our list? - Get in touch!
| Resolution | Genus level | Species level |
| Technology | Illumina short reads | Oxford Nanopore long reads |
| Target | Hypervariable region of a selected gene | Selected gene |
| Data amount you will get | From 20.000 reads/sample | From 10.000 reads/sample |

|
Along with the laboratory processing of the samples, data analysis can be ordered at the same time, which uses appropriate databases and results in graphs of taxonomic abundancies of individual taxa per sample. Results will be provided as tables/graphs and will demonstrate semi-quantified representation of microorganisms. If you already have raw data available, you can order their analysis separately. |
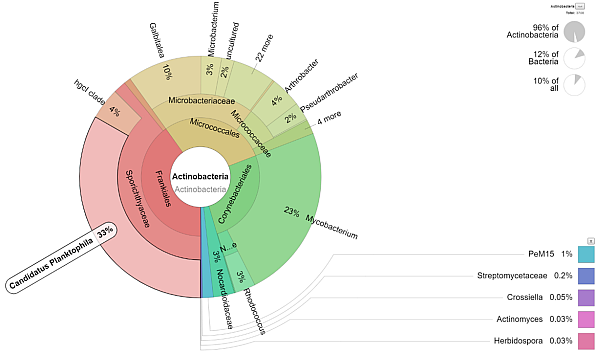 |

If you are looking for a really cheap option to sequence hundreds or thousands of amplicons at once, Tailed Amplicon Sequencing is the right choice.

Tailed-amplicon sequencing project example: Three samples (pools) containing 5 amplicons each (left) or a more sophisticated design where 15 amplicons are pooled into one sample (right). If the library contains, for example, 94 samples, 470 (left) or 1410 (right) amplicons can be sequenced in this way. The data is demultiplexed into individual samples (94 files) upon delivery. Further differentiation takes place according to the length of the product, the sequence of primers or other identifiers used.
Primer sequences and instructions can be found here.

We perform:
- RNA extraction
- RNA processing and sequencing
- Raw data quality control, trimming of bases with low quality and clipping of adaptor sequences
- Mapping to reference sequence (genome or transcriptome)
- Counting reads + Normalization
- Gene expression analysis using DESeq2 and edgeR packages
- Gene annotation and gene ontology (GO) analysis
- Evaluation and summary, data analysis report
Should you have raw data from another provider already and need only help with its analysis, we are at your disposal too.

We perform whole-exome sequencing of human and mouse DNA samples; for other organisms contact us.
Whole exome sequencing (WES) is the most effective way of studying coding regions of the genome. In our sequencing lab we utilize probe hybridization technology developed by Agilent Technologies, Inc. that has proven to be very efficient and reliable.
SureSelect XT HS enrichment system (Agilent Technologies) utilizies RNA probes to hybridize to target DNA regions. The main advantages of this technology are:
- RNA:DNA hybridization is more efficient than DNA:DNA hybridization utilized by other systems available on the market
- Sequencing libraries can be generated even using DNA from FFPE samples
- It has excellent uniformity.
- Coverage across reference sequence: 99,6% (CCDS and UCSC known genes)