Applications
Illumina technology is the so-called short read technology. It is the technology of choice if, for example, you want to analyze changes in gene expression, detect variants in (target regions of) the genome or exome, or perform shallow rather than deep screening of communities of different organisms including their functional genes. And of course in other scenarios. However, for various other applications you may rather consider long-read technology by Oxford Nanopore.
We offer and perform:
- All steps starting from project design and consulting, through DNA/RNA extraction, library preparation and sequencing using state-of-the-art instruments to standard or customized data analysis pipelines
- Solutions for large or small scale NGS projects – ShareSeq data packages
- Strict quality control on the level of the sample, the library and the data, using workflows optimized for speed and accuracy
- On-line ordering, results reporting and data delivery – all available 24/7
- Courses or workshops, also tailored to needs of your team

Illumina sequencing service overview:

When ordering Illumina sequencing you can choose one of the following options:
| You order | Sequencing by lane / flowcell | ShareSeq data package |
| What is the service suitable for? | Rather larger projects with a lower price per unit of data (eg one million reads) | Rather small to medium projects with a lower total cost |
| Available sequencing settings | Paired-end 150b, Paired-end 250b | |
| Data amount | 375 Gb and more | 1 Gb and more |
| Guarantee | The amount of data output is approximate. | If library prep is being made in our lab, we guarantee the amount of data specified for ShareSeq data packages! |
| Library preparation | We or you | |
| Library type | No limits | |
| Adapters | Compatible with standard Illumina sequencing primers. | |
| Indexes | Exclusively dual indexes. If you prepare libraries yourself and do not want / cannot use dual indexes, always contact us first. | |
| Outputs | Standard (commonly used data formats etc.). If you order a lane or flowcell, you can also get non-demultiplexed data from us (not possible with ShareSeq data packages). | |
A lane, flowcell or ShareSeq data package?
The sample processing is identical. Primarily, Share data packages are suitable for small to medium scale projects where a relatively low sequencing capacity per sample is required and it is not worth paying for full-lane sequencing. At the same time, we strongly recommend ShareSeq when you perform large and costly sequencing projects and want to run a low-cost test of, for example, ribo-depletion or other way of target enrichment for a specific genome / transcriptome fraction, reference sequence mapping rate, presence of expected organisms, etc.
For ShareSeq data packages, your samples will be analyzed along with samples from other clients. If you prepare the sequencing libraries yourself, it is absolutely critical that you provide us with accurate information on the preparation process, the indexes used, etc. This information will be requested when placing an order in our system.
Results, guarantee and data analysis
The reads obtained will be sorted according to the combination of indexes into files representing individual samples and analysis of sequencing quality indicators such as number and length of sequences, phred score,% GC, duplication level, etc. will be performed. As output you will receive data in FASTQ format divided into files according to individual samples. This Basic Data Analysis is an integral part of the sequencing.
For some types of analyzes, eg when sequencing exomes, we offer additional standard processing of results. This Advanced Data Analysis is charged based on the number of samples and is optional. If you are not interested in it, you will only receive FASTQ files (above).
Because each sequencing project has specific data analysis requirements, we offer turnkey data analysis for the needs of your project - Custom Data Analysis. Our bioinformaticians can help you with any data analysis goal. If you are interested in this service, individual consultation is required, we recommend you seek our help already at the project preparation stage.
Below is information on a few selected applications. The technological spectrum of Illumina sequencers is very extensive, in case of other applications, do not hesitate to contact us.

If you want to analyze the occurrence of microorganisms in a certain environment, metagenome sequencing is a very efficient tool of first choice. However, the disadvantage of Illumina technology compared to e.g. Oxford Nanopore technology is low resolution. Put simply, Illumina can identify microorganisms to the genus level, Oxford Nanopore (and other long read technologies) to the species level. The choice of technology is therefore determined by the aim of the experiment.
The most frequently used strategy is based on the sequencing of amplicons obtained by amplification of selected genes (long-read metagenomics) or their hypervariable regions (short-read metagenomics). Typically, these are genes for ribosomal subunits (such as 16S, 18S or ITS), but not only them. These genes or gene regions are traditionally used to identify organisms based on comparison of the obtained sequences with reference databases.
An alternative approach is shotgun sequencing of the whole sample or metatranscriptomic analysis. If you are interested in these services, please contact us.
Metagenomic analysis using amplicon sequencing
| Resolution | Genus level | Species level |
| Technology | Illumina | Oxford Nanopore |
| Target | Hypervariable region of a selected gene | Selected gene |
| Data amount you will get | From 20.000 reads/sample | From 10.000 reads/sample |
| Time to results | Approx. 4 weeks | Approx. 10 working days |
Comparison of Illumina and Oxford Nanopore outputs (example): Illumina technology is capable to discriminate down to the Bifidobacterium genus level whereas by using Oxford Nanopore technology we can identify species, e.g. Bifidobacterium breve.

What target regions do we use for taxonomic analysis?
Various hypervariable gene regions can be used for sequencing taxonomic analysis. Selection of primer combinations that we use for the amplification of these regions is continuously expanded. You can find the current available primer sets in our Sample submission sheet.
You need some other primers and cannot see them in our list? - Get in touch!
Data analysisAlong with the laboratory processing of the samples, data analysis can be ordered at the same time, which uses appropriate databases and results in graphs of taxonomic abundancies of individual taxa per sample. Results will be provided as tables/graphs and will demonstrate semi-quantified representation of microorganisms. If you already have raw data available, you can order their analysis separately. |
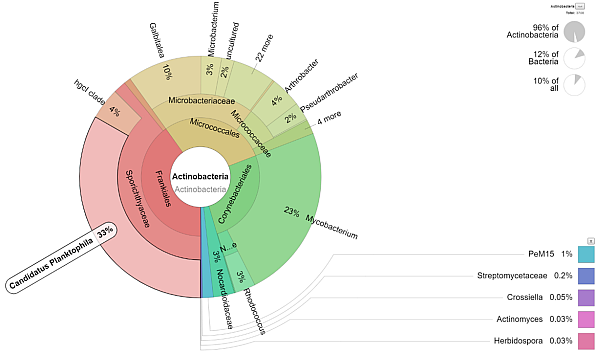 |

Differential gene expression (DGE) analysis refers to the analysis and interpretation of differences in abundance of gene transcripts within a transcriptome according to phenotype or experimental conditions. The goal of differential expression testing is to determine which genes are expressed at different levels between conditions. These genes can offer biological insight into the processes affected by the condition(s) of interest.
Because the count of genes that are differentially expressed between samples may be high, a method to understand and interpret the meaning of so many gene expression changes is needed that enables grouping of genes that belong to a particular category enriched in one sample compared to another sample. For example, if a breast cancer sample has more genes regulated that are annotated to the “cell cycle genes group” than a control sample. Grouping of genes can be performed based on their annotation to a number of sources, one of these being The Gene ontology resource (GO).
The Gene Ontology (GO) resource is the world’s largest source of information on the functions of genes. This knowledge is both human-readable and machine-readable, and is a foundation for computational analysis of large-scale molecular biology and genetics experiments in biomedical research. The GO defines concepts/classes used to describe gene function, and relationships between these concepts.
Our DGE lab & data analysis pipeline therefore includes:
- RNA extraction
- RNA processing and sequencing
- Raw data quality control, trimming of bases with low quality and clipping of adaptor sequences
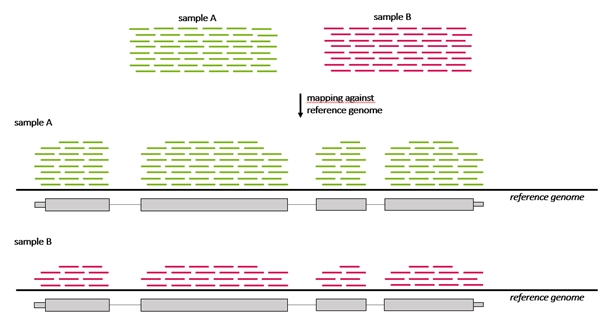
- Mapping to reference sequence (genome or transcriptome)
- Counting reads + Normalization
- Gene expression analysis using DESeq2 and edgeR packages
- Gene annotation and gene ontology (GO) analysis
- Evaluation and summary, data analysis report
Should you have raw data from another provider already and need only help with its analysis, we are at your disposal too.
Data analysis
Required read depth (data amount)
The amount of data needed per sample can be determined by the concept of depth. For example, given that the human transcriptome accounts for 3% of the human genome (3 Gb), having 90 Mb data would be 1×depth and on average cover each nucleotide of interest once. However, some genes are more highly expressed than others and some genes are rarely expressed, so even 1000×depth would only provide an even chance of sequencing a transcript that is 1 in a thousand in a cell.
The amount of data needed therefore depends on the library preparation strategy (ribodepletion, polyA selection, …), the source organism and the size of its transcriptome / genome and the genes we want to target (whether we assume high or low levels of their transcripts).
At least 30 mil. reads for DGE of human samples is generally recommended and a pilot study might be needed in case of non-model organism.
It is important to understand that a gene is declared differentially expressed if an observed difference (change in read counts) between two experimental conditions is statistically significant, that is if the difference is greater than what would be expected just due to random variation. Therefore, DGE is a statistical technique and as such must meet basic statistical requirements regarding count of samples/groups to compare. To successfully perform DGE on your data, these are the “musts”:
- Your project must be designed to have at least 2 groups of samples to compare with at least 3 biological replicates per sample/group. The smallest data set therefore is 2 groups (samples) with 3 biological replicates each = 6 data sets.
- You must specify the control and treated group.
- Genome or transcriptome reference sequence (FASTA format) and genome annotation (GFF/GTF format) must be available, preferably from public repositories such as NCBI, ENSEMBL, UCSC, etc. The reference sequence can be a genome sequence from the same organism as the source RNA or a closely related species. Reference sequences can also be closely related transcriptome sequences.
- If higher sensitivity and specificity of the experiment are needed, we recommend using RNA spike-in transcripts. Data analysis can also be performed in case of missing reference sequence or having less than 3 replicates per sample. In all these cases please contact us before ordering.
Data analysis outputs:
- Trimmed data in fastq format, multiqc report
- Aligned data in bam format, qualimap reports
- Matrix table with transcripts abundance
- Rescaled data according to the TMM normalization factors
- Expression values for all transcripts
- Volcano and MA plot in pdf format
- Count of differentially expressed genes
- Correlation heat map of each samples in pdf format
- Heat map of differentially expressed genes
- GO terms table + graphical output, web link to GO results

If you are looking for a really cheap option to sequence hundreds or thousands of DNA fragments at once, Tailed Amplicon Sequencing is the right choice. Although the name mentions amplicons, this strategy is generally suitable for any dsDNA fragments. The supplied DNA fragments must in each case carry specific overhangs as specified in the Sample Submission Guidelines. This approach leaves the initial sample preparation under your full control, allowing you to target your areas of interest while simultaneously working with the reagents of your choice, but without the need to purchase large numbers of indexed adapters.

Tailed-amplicon sequencing project example: Three samples (pools) containing 5 amplicons each (left) or a more sophisticated design where 15 amplicons are pooled into one sample (right). If the library contains, for example, 94 samples, 470 (left) or 1410 (right) amplicons can be sequenced in this way. The data is demultiplexed into individual samples (94 files) upon delivery. Further differentiation takes place according to the length of the product, the sequence of primers or other identifiers used.
Procedure
Step 1 in your lab - You PCR amplify the target regions and mix all amplicons from the same sample into one pool. Step 2 in the SEQme lab - We perform a second PCR in which we add indexes and sequencing adapters to your amplicons. Samples processed in this way are then sequenced according to your requirements - the service is flexible in terms of output.

Primer sequences and instructions for performing the first step PCR can be found here.

We perform whole-exome sequencing of human and mouse DNA samples; for other organisms contact us.
Whole exome sequencing (WES) is the most effective way of studying coding regions of the genome. In our sequencing lab we utilize probe hybridization technology developed by Agilent Technologies, Inc. that has proven to be very efficient and reliable.
SureSelect XT HS enrichment system (Agilent Technologies) utilizies RNA probes to hybridize to target DNA regions. The main advantages of this technology are:
- RNA:DNA hybridization is more efficient than DNA:DNA hybridization utilized by other systems available on the market
- Sequencing libraries can be generated even using DNA from FFPE samples
- It has excellent uniformity.
- Coverage across reference sequence: 99,6% (CCDS and UCSC known genes)
