Technologie Illumina je tzv. technologie krátkých readů. Je to technologie volby pokud chcete například analyzovat změny genové exprese, detekovat varianty v (cílových oblastech) genomu nebo exomu nebo provádět spíše mělký než hluboký screening společenstev různých organismů včetně jejich funkčních genů. A samozřejmě v dalších scénářích. Nicméně pro celou řadu aplikací může být vhodnější technologie dlouhých readů Oxford Nanopore.

- Všechny dílčí kroky nutné pro úspěch vašeho projektu, počínaje poradenstvím při jeho designu, přes izolaci DNA/RNA, přípravu sekvenačních knihoven a vlastní sekvenování na nejmodernějších přístrojích až po analýzu a interpretaci dat.
- Řešení pro velké i malé projekty NGS - datové balíčky ShareSeq
- Přísná kontrola kvality na úrovni vzorku, knihoven a dat
- On-line systém objednávání, sledování výsledků a doručování dat - vše k dispozici 24/7
- Kurzy nebo workshopy, také přizpůsobené potřebám vašeho týmu
| Objednáváte | Sekvenační dráhu nebo flowcellu | ShareSeq datový balíček |
| Na co je služba vhodná | Spíše větší projekty s nižší cenou na datovou jednotku (např. na jeden milión readů) | Spíše menší až střední projekty s nižší celkovou cenou |
| Dostupná sekvenační nastavení | Paired-end 150b, Paired-end 250b | |
| Kapacita | Od 375 Gb dat výše | Od 1 Gb dat výše |
| Záruka | Objem dat, která můžete očekávat, je určen přibližně. Většinou dodáváme větší objemy, než je uvedeno ve specifikacích. | Pokud připravujeme knihovny, pak garantujeme objem dat uvedený ve specifikacích ShareSeq datových balíčků! |
| Příprava knihoven | Náš tým nebo vy | |
| Typ knihoven | Bez omezení | |
| Adaptery | Kompatibilní se standardními sekvenačními primery Illumina. | |
| Indexy | Výhradně duální indexy. Pokud připravujete knihovny sami a nechcete/nemůžete použít duální indexy, vždy nás nejprve kontaktujte. | |
| Výstupy | Standardní (běžně používané formáty dat apod.). Pokud objednáváte dráhu nebo flowcellu, můžete od nás dostat i nedemultiplexovaná data, v případě balíčků ShareSeq to není možné. | |
Níže uvádíme informace k několika vybraným aplikacím. Technologické spektrum sekvenátorů Illumina je velmi rozsáhlé, v případě jiných aplikací nás neváhejte kontaktovat.

Chcete-li analyzovat výskyt mikroorganizmů v určitém prostředí, je sekvenování metagenomu velmi efektivním nástrojem první volby. Nejčastěji používaná strategie je založena na sekvenování amplikonů získaných amplifikací vybraných genů (metagenomika dlouhých readů) nebo jejich hypervariabilních oblastí (metagenomika krátkých readů). Typicky se jedná o geny pro ribozomální podjednotky (jako 16S, 18S nebo ITS) ale zdaleka nejen o ně. Tyto geny nebo genové oblasti jsou již tradičně využívány k identifikaci organizmů na základě srovnání získaných sekvencí s referenčními databázemi.
Aktuální dostupné cílové oblasti naleznete zde. Nenašli jste cílovou oblast, kterou potřebujete? - Kontaktujte nás!
| Rozlišení | Na úroveň rodu | Na úroveň druhu |
| Technologie | Krátké ready Illumina | Dlouhé ready Oxford Nanopore |
| Sekvenovaná oblast | Hypervariabilní úsek vybraného genu | Celý gen |
| Objem dat, který získáte | Od 20.000 readů/vzorek | Od 10.000 readů/vzorek |

|
K laboratornímu zpracování vzorků lze současně objednat i analýzu dat, která využívá vhodné databáze a jejímž výsledkem jsou grafy taxonomických četností jednotlivých kategorií a jednotlivých vzorků. Výsledky budou zpracovány v přehledné formě (tabulky, grafy) a budou demonstrovat semikvantifikované zastoupení mikroorganismů. Máte-li již hrubá data k dispozici, můžete objednat jejich analýzu samostatně. |
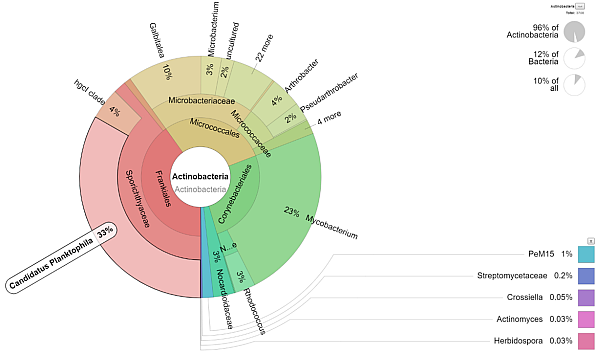 |

Pokud hledáte opravdu levnou možnost jak sekvenovat stovky nebo tisíce amplikonů najednou, je Paralelní sekvenování amplikonů tou pravou volbou.

Příklad projektu paralelního sekvenování amplikonů - tři vzorky (pooly) obsahující 5 amplikonů každý (vlevo) nebo sofistikovanější design, kdy je poolováno 15 amplikonů do jednoho vzorku (vpravo). Obsahuje-li knihovna např. 94 vzorků, lze takto sekvenovat 470 (vlevo) nebo 1410 (vpravo) amplikonů. Data jsou při dodání demultiplexována na jednotlivé vzorky (94 souborů). Další rozlišení probíhá podle délky produktu, sekvence primerů případně jiných použitých identifikátorů.
Sekvence primerů a návod na provedení najdete zde.

Provádíme:
- Izolaci RNA
- Zpracování a sekvenování vzorků RNA
- Kontrola kvality hrubých dat, trimming bází s nízkou kvalitou a odstranění sekvencí adaptérů
- Mapování na referenční sekvenci (genom nebo transkriptom)
- Počítání readů + normalizace
- Analýza genové exprese pomocí balíčků DESeq2 a edgeR
- Genová anotace a analýza genové ontologie (GO)
- Vyhodnocení a shrnutí, zpráva o analýze dat
Pokud již máte nezpracovaná data od jiného poskytovatele sekvenačních služeb a potřebujete pouze pomoci s jejich analýzou, jsme vám také k dispozici.

Standardně nabízíme celoexomové sekvenování vzorků lidské a myší DNA, pro jiné organismy nás v případě zájmu kontaktujte.
Celoexomové sekvenování (whole-exome sequencing, WES) je nejúčinnějším způsobem studia kódujících oblastí genomu. V naší sekvenační laboratoři používáme technologii hybridizace sond vyvinutou společností Agilent Technologies, Inc., která se ukázala jako velmi efektivní a spolehlivá.
Systém obohacení SureSelect XT HS (Agilent Technologies) využívá RNA sondy k hybridizaci na cílové oblasti DNA. Hlavní výhody této technologie jsou:
- Hybridizace RNA:DNA je účinnější než hybridizace DNA:DNA využívaná jinými systémy dostupnými na trhu
- Sekvenační knihovny mohou být generovány i pro DNA získanou z FFPE vzorků
- Systém má vynikající uniformitu.
- Pokrytí napříč referenční sekvencí: 99,6% (známé geny CCDS a UCSC)